Nautiyal HIMANSHU, Ph.D. Candidate 35th cycle, University of Trento, DICAM
Ph.D. degree on 25/05/2023
More than 70% of the world's energy consumption is lost as waste heat during conversion. Thermoelectric (TE) materials are a class of functional materials that can convert this waste heat into electricity. The efficiency of a TE material is measured by a dimensionless figure of merit, zT = σS2T/ (ke+ kl), where σ, S, T, ke, and kl are electrical conductivity, Seebeck Coefficient, absolute temperature, the electronic and lattice components of thermal conductivity, respectively. These transport properties show strong interdependency, which makes it challenging to maximize zT, requiring an optimal combination of S, σ, and k. The present project addresses this challenge using first-principle modelling and simulations, as a support to the experimental activity. The work primarily focuses on exploring new materials and simulating their electronic and vibrational properties, to understand the underlying principle for better thermoelectric performance.
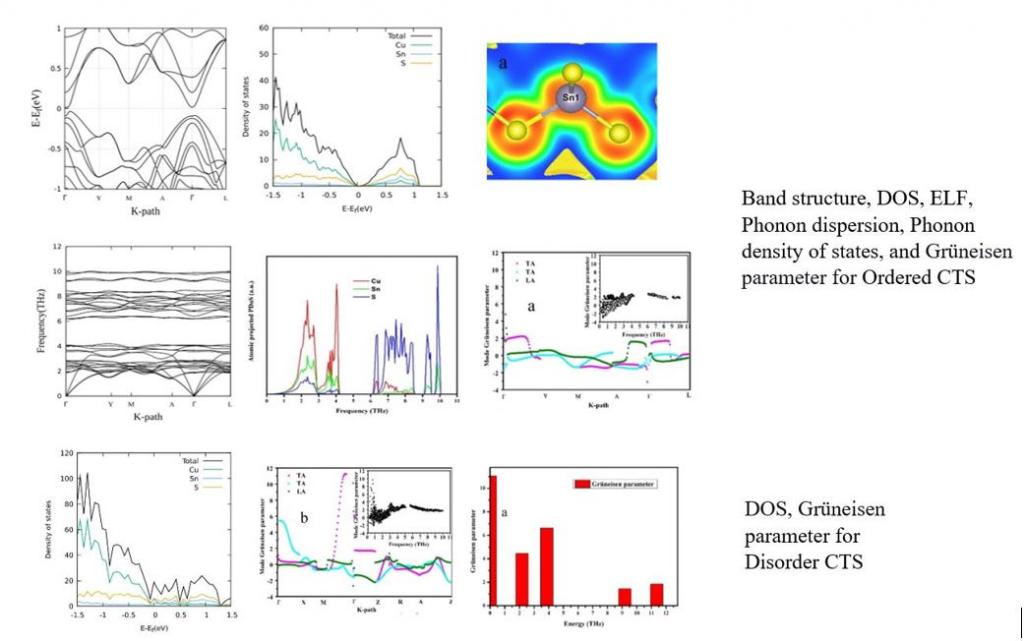
During the first year of the Ph.D., I performed first-principle calculations on Cu2SnS3 (CTS). This material, which is actively produced and tested in our laboratory, is a p-type semiconductor that exhibits several polymorphic phases, including cubic, monoclinic, tetragonal, and orthorhombic. CTS shows an interesting structural transition from disordered (cubic) to ordered (monoclinic) polymorph. Experiments performed on ordered and disordered CTS show that the disordered phase has a higher carrier concentration and an ultra-low thermal conductivity, leading to a higher figure of merit, zT.
One ordered and two disordered cells were modeled by slightly changing the partial occupancy of Cu and Sn due to stoichiometric constraints. Ab initio Density Functional Theory (DFT) and Density Functional Perturbation Theory (DFPT) simulations were performed to investigate the underlying principles of the enhanced thermoelectric performance of the disordered phase compared to the ordered one. The electronic properties were studied via the density of states (DoS), band structures, and Electron Localization Function (ELF) plots, while the vibrational properties were studied via the phonon dispersion curves, phonon density of states, group velocity, and mode-Grüneisen parameter for the anharmonicity.
Current and future work: I am presently working on simulations of electrical conductivity, Seebeck coefficient, lattice thermal conductivity, phonon lifetime, etc, to understand in detail the mechanism of electrical and thermal transport. After simulating the transport properties, I will explore new structures that could be promising and help the community, and our research group in particular, to produce new TE materials: promising candidates include inorganic perovskites and quaternary chalcogenides.
The project involves constant interactions with groups engaged in experimental activities, with national and international collaborations. Part of the work is already reported in a publication [1], while a second one is in preparation.
Bibliography
[1] K. Lohani, H. Nautiyal, N. Ataollahi, C. Fanciulli, I. Sergeev, M. Etter, & P. Scardi, “Effect of polymorphism on the thermoelectric properties of Cu2SnS3”, J. Phys. Chem. C 125, (2021) 178−188. doi: 10.1021/acs.jpcc.0c09139